filmov
tv
Complete single-cell RNAseq analysis walkthrough | Advanced introduction

Показать описание
This is a comprehensive introduction into single-cell analysis in python. I recreate the main single cell analyses from a recent Nature publication. I explain the basics of single-cell sequencing analysis and also introduce more advanced topics. I cover doublet removal, preprocessing, integration, clustering, cell identification, differential expression, gene-set enrichment, non-parametric statistical testing, single-cell gene signature scoring, plotting, and more. This tutorial is suitable for both advance and new single-cell users. I use the scanpy and SCVI packages heavily.
Notebook:
Reference:
0:00 intro
1:18 data
6:35 doublet removal
13:03 preprocessing
23:12 Clustering
27:42 Integration
39:56 label cell types
58:28 Analysis
Notebook:
Reference:
0:00 intro
1:18 data
6:35 doublet removal
13:03 preprocessing
23:12 Clustering
27:42 Integration
39:56 label cell types
58:28 Analysis
 1:18:40
1:18:40
Complete single-cell RNAseq analysis walkthrough | Advanced introduction
 2:50:44
2:50:44
W20: Single-Cell RNA-Seq Analysis with Python - Day 1
 0:05:50
0:05:50
Introduction to single-cell RNA-Seq and Seurat | Bioinformatics for beginners
 0:40:32
0:40:32
Single cell transcriptomics - Introduction to single cell RNA-seq (1 of 10)
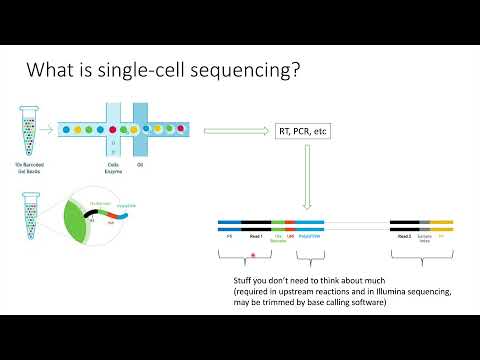 0:02:35
0:02:35
Single-cell sequencing explained in 2 minutes
 0:24:37
0:24:37
Single Cell Sequencing - Eric Chow (UCSF)
 2:38:33
2:38:33
W20: Single Cell RNA-seq with R – Day 1
 0:17:41
0:17:41
Single cell RNA sequencing overview | ScRNA seq vs Bulk seq | chemistry of ScRNA seq |Bio Techniques
 0:09:45
0:09:45
InTraSeq™ Single Cell Analysis: Multimodal, Simultaneous Measurement of RNA and Proteins
 0:07:33
0:07:33
Introduction to ScRNA-seq Data Analysis
 1:09:30
1:09:30
325: Transcriptomics Unveiled – An In-Depth Exploration of Single Cell RNASeq Analysis using python...
 0:26:09
0:26:09
A beginner's bioinformatics guide for single-cell RNAseq data analysis
 0:12:24
0:12:24
The Beginner's guide to bulk RNA sequencing vs single-cell RNA Sequencing
 0:36:18
0:36:18
How to analyze single-cell RNA-Seq data in R | Detailed Seurat Workflow Tutorial
 0:51:12
0:51:12
Rafael Irizarry, Probabilistic Gene Expression Signatures for Single Cell RNA seq Data
 1:52:12
1:52:12
W20: Single-Cell RNA-seq with R – Day 3
 0:54:15
0:54:15
GDC Single Cell RNA-Seq Support – September 27, 2021 GDC Monthly Webinar
 1:17:40
1:17:40
Anna Cuomo & Ximena Ibarra - Single-cell tutorial [6/6]: scRNA seq data analysis diff. expr.
 0:41:17
0:41:17
Life cell by cell: Introduction to Single Cell Expression Atlas
 0:47:44
0:47:44
How to Analyze Single Cell RNA Seq Data - Point, Click, Done
 0:15:26
0:15:26
Introduction to scRNA-seq data analysis
 0:02:39
0:02:39
Single Cell Gene Expression Protocol v3.1 | Assemble Chromium Next GEM Chip G
 0:27:12
0:27:12
Practical Considerations and Best Practices for Single Cell and Spatial Transcriptomics
 0:58:45
0:58:45
Introduction of R programming 9 Single cell RNA seq data analysis
Комментарии