filmov
tv
X-ray crystallography and cryo-em (cryo-electron microscopy): an overview & comparison
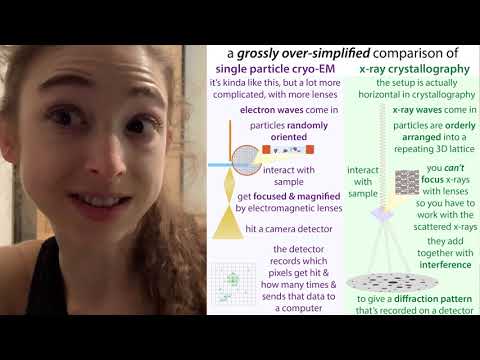
Показать описание
Dying to know “What’s the deal with cryo?” Cryo-Electron Microscopy (CryoEM) has transformed the structural biology field in recent years, and even garnered some wider fame when Jacques Dubochet, Joachim Frank and Richard Henderson won the Nobel Prize for it in 2017. So what is it? How does it compare the the “traditional” x-ray crystallography? And is crystallography dead? (hint: NO!) Each of these techniques has pros and cons. So let’s take a look at what’s going on. Starting with “What is structural biology anyway?” (I didn’t really even hear this term until late in undergrad and people kinda just expected me to know and never really explained, so here’s my attempt at it…)
“Structural biology” is a sub-field of biology that’s focused on figuring out what molecules “look like” and how their shape is connected to their function. A little more on this later, but for now just think of a silver fork and spoon. They’re made up of the same material so just figuring out “hey these are made of silver” isn’t going to tell you much about what they do (but feel free to pat yourself on the back - I’m all for celebrating little wins!). If you play around with them blindly (and carefully), you might be able to figure out that you can eat soup with a spoon but not a fork but it’s easier to stab and eat pasta noodles with your fork. And if you can see them, it’s easy to see how their distinct shapes allow them to perform differently at these tasks.
Structural biology works to connect the “what it does” to the “what it looks like” to help determine “how it does it” and it uses techniques including X-ray crystallography and cryo-EM to get looks, combined with biochemical & biophysical techniques (measuring activity, binding, etc.) to further investigate the functional importance of different parts that are seen (though sometimes this functional stuff is discovered first but makes a lot more sense once you can see it). It’s kinda like how if you make a hole in the spoon-y part of the spoon it makes your soup drip before it gets to your lip & if you see the spoon shape you can predict where hole-making might make you dribble. But, if you make a hole in the handle you might not know.
And speaking of things you might not know, let’s discuss “cryo” - “cryo” (which my computer insists on de-correcting to cry, so apologies if I don’t catch all the “corrections”) is short for “cryogenic” meaning really really cold (which we need to keep the molecules safe(ish) and still(ish)). And when structural biologists say “cryo” they’re usually referring to single particle cryo-electron microscopy (cryoEM) as opposed to things like cryo-tomography, which looks at “slabs” like sections of cells or tissues.
You might sometimes hear it called single *molecule* cryoEM, but the things you’re looking at don’t have to be individual molecules. A molecule is something where all the atoms (the individual carbons, hydrogens, etc.) are connected through strong covalent bonds (like one protein chain) as opposed to “complexes” where multiple molecules can interact with one another through weaker interactions (like multiple protein subunits and RNA pieces working together to make a ribosome). So “particles” is a broader term that encompasses single proteins, multi-protein complexes, protein/DNA complexes, etc. - any sort of individual “particles” in solution (i.e. each particle surrounded by its own full water coat).
Disclaimer - even though I earned my PhD right down the hall from a top-of-the-line cryo-electron microscope, I personally don’t use cryo-EM, but have done some crystallography and will talk more about this at the end.
So imagine you have a bunch of copies of one of these particles. And you want to figure out what they look like…
If you want to look at something small, your first thought might be - let’s use a microscope. Microscopes make things look bigger by taking advantage of the wave properties of light (visible light is a form of ElectroMagnetic Radiation (EMR) - which can be thought of as packets of energy called photons traveling as waves). When a wave interacts with things, the wave’s path can get altered. So a visible microscope can shine light through something and have that thing alter the light waves’ paths. Thanks to that altering, you now you have a bunch of waves “out of phase” (out of step with one-another “peak-wise”). So the signal from your thing is kinda jumbled.
finished in comments
“Structural biology” is a sub-field of biology that’s focused on figuring out what molecules “look like” and how their shape is connected to their function. A little more on this later, but for now just think of a silver fork and spoon. They’re made up of the same material so just figuring out “hey these are made of silver” isn’t going to tell you much about what they do (but feel free to pat yourself on the back - I’m all for celebrating little wins!). If you play around with them blindly (and carefully), you might be able to figure out that you can eat soup with a spoon but not a fork but it’s easier to stab and eat pasta noodles with your fork. And if you can see them, it’s easy to see how their distinct shapes allow them to perform differently at these tasks.
Structural biology works to connect the “what it does” to the “what it looks like” to help determine “how it does it” and it uses techniques including X-ray crystallography and cryo-EM to get looks, combined with biochemical & biophysical techniques (measuring activity, binding, etc.) to further investigate the functional importance of different parts that are seen (though sometimes this functional stuff is discovered first but makes a lot more sense once you can see it). It’s kinda like how if you make a hole in the spoon-y part of the spoon it makes your soup drip before it gets to your lip & if you see the spoon shape you can predict where hole-making might make you dribble. But, if you make a hole in the handle you might not know.
And speaking of things you might not know, let’s discuss “cryo” - “cryo” (which my computer insists on de-correcting to cry, so apologies if I don’t catch all the “corrections”) is short for “cryogenic” meaning really really cold (which we need to keep the molecules safe(ish) and still(ish)). And when structural biologists say “cryo” they’re usually referring to single particle cryo-electron microscopy (cryoEM) as opposed to things like cryo-tomography, which looks at “slabs” like sections of cells or tissues.
You might sometimes hear it called single *molecule* cryoEM, but the things you’re looking at don’t have to be individual molecules. A molecule is something where all the atoms (the individual carbons, hydrogens, etc.) are connected through strong covalent bonds (like one protein chain) as opposed to “complexes” where multiple molecules can interact with one another through weaker interactions (like multiple protein subunits and RNA pieces working together to make a ribosome). So “particles” is a broader term that encompasses single proteins, multi-protein complexes, protein/DNA complexes, etc. - any sort of individual “particles” in solution (i.e. each particle surrounded by its own full water coat).
Disclaimer - even though I earned my PhD right down the hall from a top-of-the-line cryo-electron microscope, I personally don’t use cryo-EM, but have done some crystallography and will talk more about this at the end.
So imagine you have a bunch of copies of one of these particles. And you want to figure out what they look like…
If you want to look at something small, your first thought might be - let’s use a microscope. Microscopes make things look bigger by taking advantage of the wave properties of light (visible light is a form of ElectroMagnetic Radiation (EMR) - which can be thought of as packets of energy called photons traveling as waves). When a wave interacts with things, the wave’s path can get altered. So a visible microscope can shine light through something and have that thing alter the light waves’ paths. Thanks to that altering, you now you have a bunch of waves “out of phase” (out of step with one-another “peak-wise”). So the signal from your thing is kinda jumbled.
finished in comments
 0:02:37
0:02:37
What is Cryo-Electron Microscopy (Cryo-EM)?
X-ray crystallography and cryo-em (cryo-electron microscopy): an overview & comparison
 0:00:29
0:00:29
Methods for Determining Atomic Structures: X-ray Crystallography (from PDB-101)
 0:34:07
0:34:07
Yifan Cheng (UCSF & HHMI) 1: Single Particle Cryo-EM
 0:39:52
0:39:52
Channelopathies resolved via cryo-EM and X-ray crystallography
 0:07:27
0:07:27
Lecture 01, concept 18: Protein structure determination - X-ray crystallography & Cryo-EM
 0:37:27
0:37:27
Modeling of protein structural heterogeneity in X-ray Crystallography and CryoEM
 1:02:38
1:02:38
March 28, 2024: Prof Wolfgang Peti, University of Connecticut Health Center
 0:03:47
0:03:47
Determining 3D structures of macromolecular complexes using Cryo-EM to understand function
 0:01:22
0:01:22
Cryo-EM Animation
 0:05:00
0:05:00
The 2017 Nobel Prize in Chemistry: Cryo-electron microscopy explained
 0:28:44
0:28:44
From Electron Crystallography to Single Particle cryoEM
 0:15:47
0:15:47
Comparison of Single crystal X-ray diffraction, NMR and Cryo-EM
 2:42:26
2:42:26
Lecture 5 - Cryo-Electron Microscopy - How TEM works
 0:01:11
0:01:11
David shooting X-rays at a protein crystal
 0:02:11
0:02:11
Cryo-EM: Structural insights for everyone
 1:00:47
1:00:47
Using Cryo-Electron Microscopy for Structural Biology | EMSL LEARN Webinar Series
 0:28:44
0:28:44
Nobel Lecture: Richard Henderson, Nobel Prize in Chemistry 2017
 0:30:34
0:30:34
CCP4 SW2020 - Densities in X-ray crystallography and electron microscopy - speaker: Garib Murshudov
 0:04:31
0:04:31
X Ray Crystallography & Cryo-EM in the Fight Against COVID 19 - Patrick S
 0:02:59
0:02:59
The New York Structural Biology Center
 0:02:38
0:02:38
Using cryo electron microscopy and tomography to advance muscle research at the Max Planck Institute
 0:39:10
0:39:10
CU School of Medicine Cryo-EM Structural Biology Shared Resource Facility
 0:49:27
0:49:27
Dr Richard Henderson - Using electron microscopy to understand the molecules of life
Комментарии