filmov
tv
Analyzing Public Datasets 2: Finding the Data

Показать описание
In this new series, we'll learn how to access and analyze public datasets resulting from next-generation sequencing techniques such as Illumina and 454.
This video shows how to find a sample dataset, upload it to Galaxy, and process it for alignment.
This video shows how to find a sample dataset, upload it to Galaxy, and process it for alignment.
 0:08:59
0:08:59
Analyzing Public Datasets 2: Finding the Data
 0:07:44
0:07:44
Best Places to Find Datasets for Your Projects
 0:09:25
0:09:25
Analyzing Public Datasets 6: From start to finish... a TopHat alignment
 0:06:23
0:06:23
Analyzing Public Datasets 4: Viewing the Alignment
 0:00:05
0:00:05
Master Data Analyst in 2024 with This Proven Roadmap
 0:00:05
0:00:05
5 Essential SQL Concepts to Ace Your Data Analysis Interview
 0:04:12
0:04:12
Analyzing Public Datasets 3: Performing an Alignment
 0:00:39
0:00:39
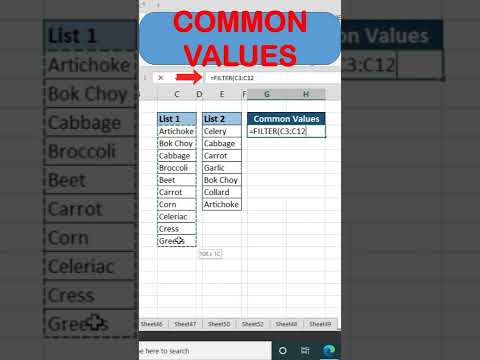
Find Common Values From Two List In Excel @BrainUpp
 0:10:20
0:10:20
A Beginners Guide To The Data Analysis Process
 0:03:38
0:03:38
Analyzing Public Datasets 1: Introduction to Galaxy
 0:05:02
0:05:02
Analyzing Public Datasets 7: An Introduction to IGV (genomic)
 0:00:28
0:00:28
2 websites for finding ML Datasets! 📈🤖 #programming #coding #machinelearning
 0:08:54
0:08:54
Analyzing Public Datasets 5: Introduction to IGV (cDNA)
 0:00:14
0:00:14
Life as a Data Analyst #shorts
 0:05:47
0:05:47
10 Free Dataset Resources for Your Next Project!
 0:11:21
0:11:21
Loading, Viewing, working with an R dataset (basics)
 0:00:27
0:00:27
Don’t Become a Data Scientist If
 0:00:54
0:00:54
My 3 go-to places to find interesting real-life datasets
 0:00:31
0:00:31
How to compare two lists to find missing values in excel - Excel Tips and Tricks
 0:00:15
0:00:15
Real Time Power BI Project, Blinkit Analysis #powerbi #powerbidashboard #dataanalyst
 0:33:07
0:33:07
Explore data from chronic liver disease and find other related public datasets.
 0:00:25
0:00:25
🔥Salary of a Data Scientist | How Much Do Data Scientists Make? | Intellipaat #Shorts #DataScientist...
 0:01:38
0:01:38
Google Cloud Public Datasets Program Overview
 0:00:14
0:00:14
5 FREE websites with datasets for your data projects: 1
Комментарии